If you've just heard the term "congenital adrenal hyperplasia" — maybe from a doctor, a newborn screening result, or a search you did late at night — you're probably looking for an easy-to-digest explanation. Here's what you need to know about classical CAH.
What CAH is — and what it isn't
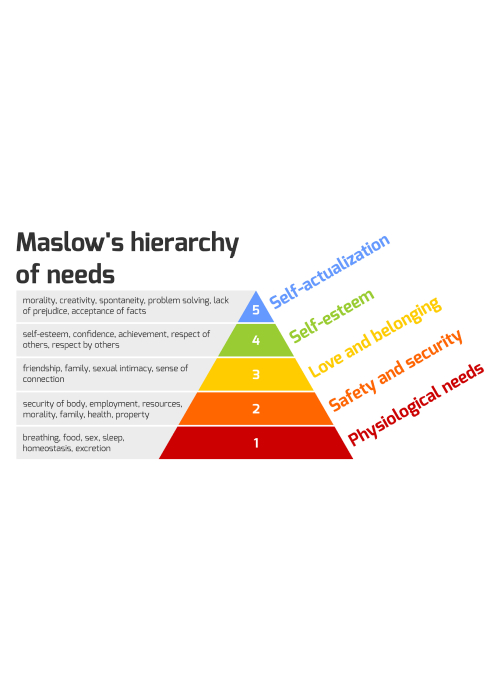
Congenital adrenal hyperplasia, or CAH, is a genetic condition that affects the adrenal glands: two small glands that sit on top of your kidneys. These glands make hormones your body needs to function, including cortisol (which helps you handle stress and illness), aldosterone (which regulates salt and fluid balance), and androgens (sex hormones).
In CAH, the body is missing an enzyme needed to make these hormones correctly. In most cases, the missing enzyme is called 21-hydoxylase, which is produced by a gene called CYP21A2. When this enzyme doesn't work properly, the body can't make enough cortisol or aldosterone. Instead, the body produces too many androgens, like testosterone.
CAH is not a disease you develop later in life. It's present from birth, which is why the word "congenital" is in the name. It's also inherited, meaning it runs in families.
{{cah-start}}
Classical vs. non-classical: what's the difference?
CAH comes in different forms. Classical CAH is the more severe form and is present from birth. Non-classical CAH is a milder form that may not be diagnosed until later in childhood or adulthood. This article focuses on classical CAH.
Classical CAH itself has two subtypes. The salt-wasting form is the most common and most serious. The body can't make enough aldosterone, which means it can't hold onto salt properly. Without treatment, this can cause a life-threatening adrenal crisis in the first few weeks of life. The simple virilizing form is less severe — the body makes enough aldosterone, but still produces too many androgens, which affects physical development.
How common is classical CAH?
Classical CAH occurs in roughly 1 in every 15,000 births. It affects boys and girls equally, though the effects look different depending on sex. It is more common in Indigenous peoples, particularly Yup'ik communities, in the United States.
What does it look like at birth?
In infants assigned female at birth, the most visible sign is often ambiguous genitalia — the external sex organs may appear different from typical female anatomy due to excess androgen exposure before birth. In infants assigned male at birth with classical CAH typically look “typical” at birth, which is one reason newborn screening is so important — without a blood test, a salt-wasting crisis may be the first sign something is wrong.
How is CAH diagnosed?
In the United States, screening for classical CAH is standard for newborn babies. A few drops of blood taken from a baby's heel in the first days of life are tested for elevated levels of a hormone called 17-hydroxyprogesterone (17-OHP), which builds up when the 21-hydroxylase enzyme is missing. Early detection for newborns is important – one large research study from 2025 showed that newborn screening for classical CAH is effective at preventing life-threatening adrenal crises. Getting test results early, and working closely with a healthcare provider after receiving a positive result, is important for long-term care.
What happens after diagnosis?
Classical CAH is a lifelong condition. There is no cure, but it is treatable. People with classical CAH take hormone replacement medications every day — typically hydrocortisone to replace cortisol, and fludrocortisone (for salt-wasting CAH) to replace aldosterone. With the right treatment and monitoring, most people with classical CAH can live full, healthy lives.
{{cah-end}}
Why research still matters — and how patients can help.
Even with decades of treatment experience, there is still a lot researchers don't fully understand about classical CAH. How does lifelong hormone replacement affect the body over time? What does long-term health really look like across thousands of patients with different subtypes? These are questions that observational research is uniquely positioned to answer — by following real patients through their real lives, not just in clinical trials.
If you or your child has classical CAH, your medical history is a meaningful contribution to that effort.
Learn more about participating in CAH research